

Abstract
Chronic myeloid leukemia (CML) is a clonal myeloproliferative disorder characterized by Philadelphia chromosome and its molecular counterpart, the BCR-ABL1 fusion gene. Discontinuation of tyrosine kinase inhibitors (TKIs) therapy after achieving a persistent deep molecular response (DMR) is an urgently-needed treatment goal for patients and it was written in NCCN guideline Version 2.2017 for CML. Indeed, various studies have confirmed the feasibility of stopping TKIs therapy in many regions. However, increasing evidence demonstrated that TKIs are unable to eliminate quiescent leukemic stem cells (LSCs), which lead to treatment resistance or relapse after discontinuation of TKIs treatment. Multiple works have confirmed that approximately 50-60% of DMR patients stopping TKIs will lose their response and require retreatment. The patients who will lose the treatment-free remission (TFR) remains unknown currently.
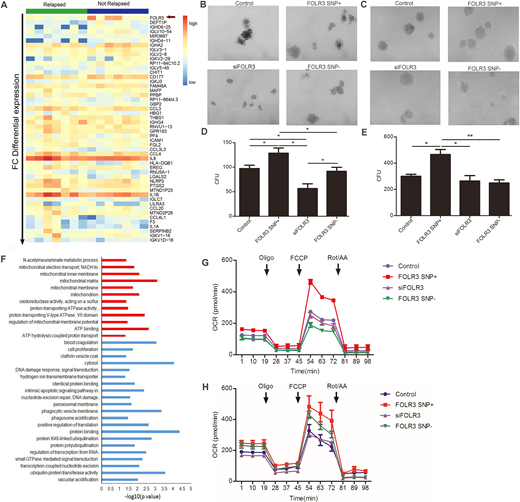
We selected bone marrow of 14 patients (matched 7 relapse and 7 non-relapse patients) who discontinued TKIs therapy in our center for transcriptome analysis to discover the differences between relapse and non-relapse patients. Bioinformatics indicated that folate receptor 3 (FOLR3) was highly expressed in non-relapse patients and only in the non-relapsers (Figure 1A). Further work found these samples all carried a common SNP mutation. Then we respectively transferred newly diagnosed chronic phase (CP) CML patients derived bone marrow CD34+ cells and CML K562 cells with lentiviral vectors containing FOLR3 SNP, FOLR3 SNP shRNA, and wild type FOLR3. The result indicated that FOLR3 SNP significantly promoted the clonogenicity of CD34+/K562 cells, while loss of FOLR3 SNP hindered cell differentiation (Figure 1B and D are data of CD34, Figure 1C and E are data of K562). K562 subcutaneous tumor formation in balb/c node mice confirmed that tumors weight and volume of FOLR3 SNP shRNA group were higher than control. While, small animal PET scanning showed that maximum standardized uptake value (SUVmax) of 18F-FDG of FOLR3 SNP+ group was higher than the rest. Experiments in vivo and vitro synergistically proved FOLR3 SNP promoted tumor cell differentiation, delayed tumor growth.
To better understand how FOLR3 SNP promote tumor cell differentiation and delay tumor growth, we performed second transcriptome analysis. Consistently, both in FOLR3 SNP+ CD34+ and K562 cells, enrichment analysis revealed that differentially expressed genes were enriched in mitochondria associated Gene Ontology (GO) biological process (Figure F), in which mitochondria complex V matched genes were most significant, such as ATP5 family. In vitro, we demonstrated that ATP syntheses, maximal respiration and spare respiratory capacity of FOLR3 SNP+ CD34+/K562 cells were significantly higher than control and shFOLR3 counterpart through seahorse XF cell mito stress test (Figure G and H refer to CD34 and K562, separately). Electron microscope also exhibited an increase of mitochondrial in FOLR3 SNP+ cells.
Mitochondria play an essential role in energy generation, cell signalling, differentiation, death and senescence in eukaryotic cells. We detected a series of genes related to aging, cell cycle and mitochondria unfolded protein response. The results showed that cell cycle kinase such as CDK4 decreased in FOLR3 SNP+ CD34+/K562 cells. On the other hand, senescence associated genes seemed increased. In conclusion, we highlighted the connection of FOLR3 and post-cessation relapse. FOLR3 SNP could be an indicator of TFR. Its internal mechanism might be the mitochondrion activation induced aging of residual leukemia cells.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal